Genome sequence of the soybean fungal pathogen, Cercospora kikuchii
Related Research Project
Resilient crops
Description
Diseases caused by Cercospora spp. are threats to soybean production in South American countries such as Argentina. Cercospora kikuchii (Tak. Matsumoto & Tomoy.) M. W. Gardner is a pathogen of Cercospora leaf blight and purple seed stain in soybean. The symptoms appear on several parts of soybean plant, for instance, leaves, petioles, and seeds. Fungicide is one of the main management methods against this devastating disease. However, it was reported that some agrochemicals are losing effectiveness against the pathogen. High-quality genomic information of C. kikuchii can provide fundamental insights toward understanding the disease, and such studies will contribute to designing a molecular diagnosis method for the disease.
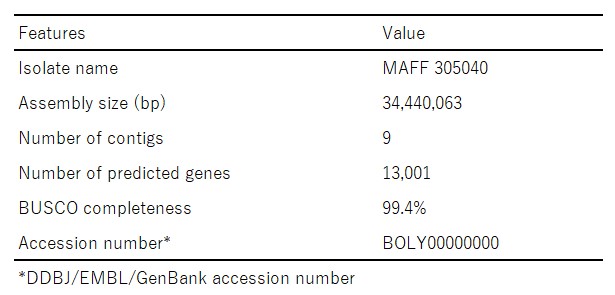
We selected the C. kikuchii isolate MAFF 305040 for this research. This was isolated in Japan and deposited to the Genebank of the National Agriculture and Food Research Organization (Tsukuba, Ibaraki, Japan). Sufficient depth of genome sequencing generated a high-quality genome assembly of this pathogen. The final genome assembly contained nine contigs comprising 34.44 Mb (Table 1, Fig. 1). The number of genes predicted from the genome assembly was 13,001 (Table 1). Completeness of the dataset was estimated using BUSCO (Table 1). From the predicted coding sequences, candidates for pathogenicity-related genes, namely, effector genes, secondary metabolite gene clusters, and genes of carbohydrate-active enzymes (CAZymes) were selected (Fig. 1).
The genome sequence was deposited in public databases (Table 1). The data can be used as a source in C. kikuchii genomic studies. Coding regions and functions of encoding proteins in this genome sequence should be validated since these were predicted from the assembled genomic sequence based on the database of genes/proteins of other organisms.
Figure, table
-
Table 1. Description of the genome assembly
-
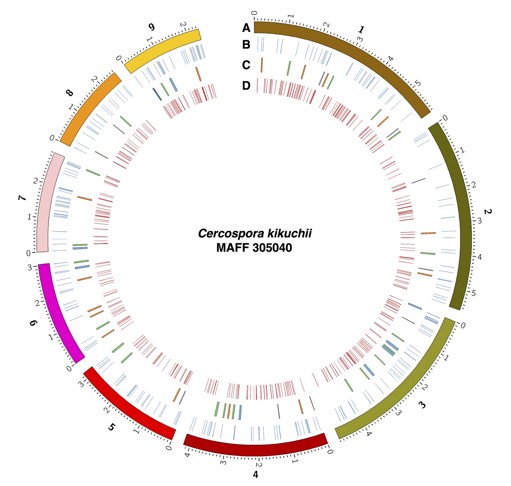
Fig. 1. Graphical summary of the genome assembly
Circos plot of the genome assembly. Track A indicates nine contigs of the genome assembly. Minor ticks indicate 0.1 Mb. Positions of the predicted pathogenicity-related genes, namely, effector candidates (B), secondary metabolite gene clusters (C), and CAZyme genes (D) are also shown.Figure and table reprinted/modified with permission from Kashiwa and Suzuki (2021).


- Classification
-
Research
- Research project
- Program name
- Term of research
-
FY 2017–2021
- Responsible researcher
-
Kashiwa Takeshi ( Biological Resources and Post-harvest Division )
KAKEN Researcher No.: 60766400Suzuki Tomohiro ( Utsunomiya University )
KAKEN Researcher No.: 10649601 - ほか
- Publication, etc.
-
Kashiwa and Suzuki (2021) G3: Genes|Genomes|Genetics, 11 (10), jkab277.https://doi.org/10.1093/g3journal/jkab277
- Japanese PDF
-
2021_B04_ja.pdf299.42 KB
- English PDF
-
2021_B04_en.pdf245.76 KB
- Poster PDF
-
2021_B04_poster.pdf221.18 KB
* Affiliation at the time of implementation of the study.