Possibility to introduce genomic selection into tree breeding for important timber species distributed in the tropical rainforests of Southeast Asia
Country
Malaysia
Description
In Southeast Asian rainforests, commonly adopted logging systems allow only trees larger than the regulated cutting limit to be selectively felled, with the next harvest expected after the forests have naturally recovered (35-year rotation in Indonesia). However, poor recovery and a decline in production levels have caused problems in second and further harvesting. Indonesia, which has the largest tropical rainforest in Southeast Asia, is implementing a system that restores productivity by artificial planting after logging, but the seedlings have not been genetically improved. Thus, this research aimed to clarify how genetically improved seedlings greatly contribute to the recovery of tropical rainforests and the improvement of productivity.In recent years, advances in next-generation sequencing have made it possible to detect genome-wide DNA polymorphisms from a large number of individuals. This technology allows the development of a genomic prediction model that can estimate phenotypes from DNA polymorphisms, which can be used for the selection of superior progenies in the next generation (genomic selection). Conventional forest tree breeding requires a long period of time to evaluate phenotypes due to their longevity, but predicting future phenotypes from DNA polymorphisms at seedling stage can significantly shorten the breeding cycle. Therefore, to restore the rainforest and improve productivity with superior seedlings, we clarified the possibility of genomic selection for dipterocarp species, which are the dominant tree species in the rainforest and provide various ecosystem services including timber production.
Genome-wide DNA polymorphisms (5,900 loci) were obtained from 356 progenies derived from open-pollinated mating of 77 mother trees of Shorea platyclados, one of the main dipterocarp timber species, which were planted in a progeny trial at a forest concession area in central Kalimantan. Linkage disequilibrium over long physical distances showed that the progenies were suitable for genome-wide association studies (GWAS) and genomic predictions. GWAS revealed that a locus showed significant association with tree height before thinning (8th year), but did not exhibit significant association with tree height after thinning (11th year), trunk diameter, tree shape, and wood density, which suggest that these traits are regulated by many weakly effective genes. Selection of informative loci based on GWAS improved genome heritability, which represented that phenotypic variance was well explained by the genotype of the selective loci (Table 1). Furthermore, the genomic heritability of growth-related traits is higher than that of tree shapes and wood density. Therefore, genomic selection is particularly effective for breeding fast-growing individuals.
We identified that deep learning and increasing the numbers of analyzed individuals and loci contribute to improving the accuracy of the genomic prediction model. Using the developed genome prediction model, selection of seedlings for growth-related traits will lead to a significant shortening of the breeding cycle. However, our results suggest that these traits are regulated by many weakly regulated genes, hence are not suitable for marker-assisted selection.
Figure, table
-
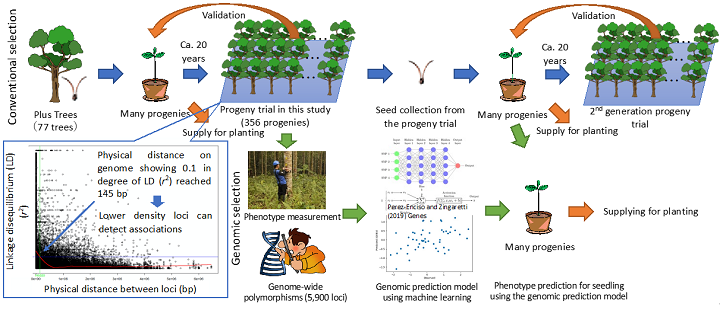
Fig. 1. Genetic characteristics of the progeny trial in this study and genomic selection
Conventional selection requires about 20 years for validation. The genomic prediction model can shorten this process, which in turn can shorten the breeding cycle.
-
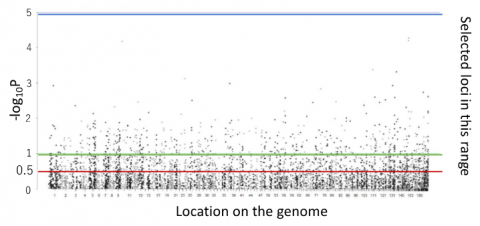
Fig 2. Manhattan plot showing association between tree height after thinning and genotype of each loci
GWAS on all measured traits showed no significant association except for a significant locus on tree height before thinning.
-
Table 1. Genomic heritability estimated from genotypes
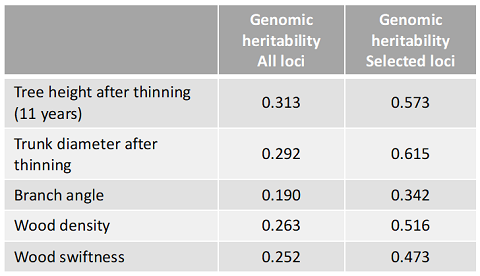
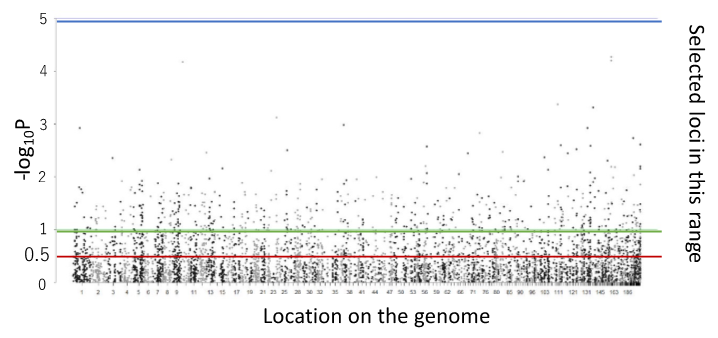
Genomic heritabilities on growth (tree height and trunk diameter) were improved when the loci were selected using a –log10P value threshold of 0.5 (red line in Fig. 2), which indicated that there are many associated genes with weak effect.


- Affiliation
-
Japan International Research Center for Agricultural Sciences Forestry Division
- Classification
-
Research
- Research project
- Program name
- Term of research
-
FY2020(FY2016~FY2020)
- Responsible researcher
-
Tani Naoki ( Forestry Division )
ORCID ID0000-0001-9878-6352KAKEN Researcher No.: 90343798Suwa Rempei ( Forestry Division )
ORCID ID0000-0003-4401-5581KAKEN Researcher No.: 40535986Sawitri ( Gadjah Mada University )
ORCID ID0009-0000-2925-7027Na'iem Mohammad ( Gadjah Mada University )
Widiyatno ( Gadjah Mada University )
ORCID ID0000-0002-6611-5009Indrioko Sapto ( Gadjah Mada University )
ORCID ID0000-0003-1828-469XUchiyama Kentaro ( Forestry and Forest Products Research Institute )
Tsumura Yoshihiko ( University of Tsukuba )
ORCID ID0000-0002-9844-0914KAKEN Researcher No.: 20353774 - ほか
- Publication, etc.
-
Sawitri et al. (2020) Forests, 11 (2):239https://doi.org/10.3390/f11020239
- Japanese PDF
-
2020_C06_A4_ja.pdf837.65 KB
2020_C06_A3_ja.pdf785.12 KB
- English PDF
-
2020_C06_A4_en.pdf835 KB
2020_C06_A3_en.pdf801.08 KB
- Poster PDF
-
2020_C06_poster.pdf536.86 KB
* Affiliation at the time of implementation of the study.